The wealth of human tumor genomic data and mechanistic understanding of drivers of disease progression allow for a more refined stratification of tumor subtypes and disease states. Precision medicine targeted therapies are based on an enhanced understanding of tumor biology, such as genomic mutations or changes in protein expression that drive cell proliferation. Patient tumors are assessed to identify a requisite biomarker and inform clinical therapy, with the goal of driving higher response rates.
G enetic sequencing and protein profiling are increasingly available to patients, clinicians, and researchers, for example, through broad-based tumor-profiling diagnostic panels. These tumor-profiling panels are dynamic – being updated by diagnostic companies to reflect an increasing number of targets – adding breadth and depth of content for characterizing patient tumors.
With genetic screening techniques continuing to improve and gain in clinical practice, we believe that precision medicine approaches should result in the discovery and development of more effective targeted therapies.

Synthetic Lethality is an emerging area within precision medicine oncology, presenting an opportunity to deliver the next generation of targeted therapies for patients. Advancements in molecular biology research, including genetic knockdown and editing capabilities, are enabling the identification and validation of novel biological targets. Programs in Synthetic Lethality represent exciting opportunities with the potential to be broadly impactful for patients.
William R. Sellers, M.D.
IDEAYA Scientific Advisory Board

IDEAYA Synthetic Lethality Platform
IDEAYA has established a fully-integrated platform that enables synthetic lethality target and biomarker discovery, drug discovery, functional genomic and pharmacological validation, translational research and opportunity expansion.

Synthetic Lethality
Synthetic lethality is emerging as an important therapeutic paradigm in the treatment of cancer. It was first defined by Calvin Bridges in 1922 based on the observation that certain combinations of gene mutations resulted in cell lethality despite the fact the single mutations in either gene were viable.
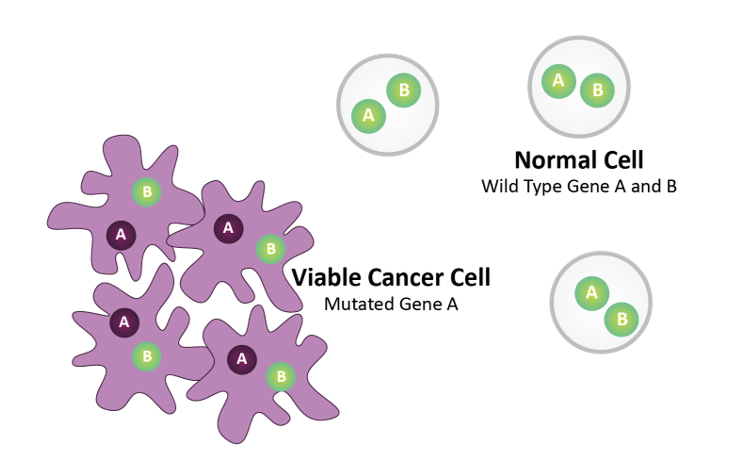
Mutated, amplified, and deleted genes in cancer have been catalogued, and a subset of historically undruggable targets can potentially be pursued indirectly, based on the concept of synthetic lethality. For example, cancer cells with loss of function mutations in a first gene “A” may have susceptibility to pharmacological inhibition of its synthetic lethal partner, the protein product of gene “B”. Cancer cells often contain genetic changes that lead to alterations in pathways such as DNA repair and metabolism. These changes endow the cancer cells with certain properties such as the ability to replicate by bypassing normal control mechanisms. However, removing these important regulators of cell function may also make these cancer cells more dependent on backup pathways that can then be targeted to achieve a therapeutic effect.
IDEAYA is prosecuting a novel set of drug targets selected through consideration of the robustness and conservation of synthetic lethality interactions across different organisms and in human tumor cells, disease relevance of drug target and prevalent loss-of-function mutation in a synthetic lethality partner gene, and ability to drug the target with a therapeutic.
Pre-SL Inhibitor Treatment
Gene A = Biomarker (e.g., BRCA)
Gene B = Drug Target (e.g., PARP)
Normal cells harbor wild type Gene A and Gene B. Tumor cells harbor mutated Gene A.
Post-SL Inhibitor Treatment
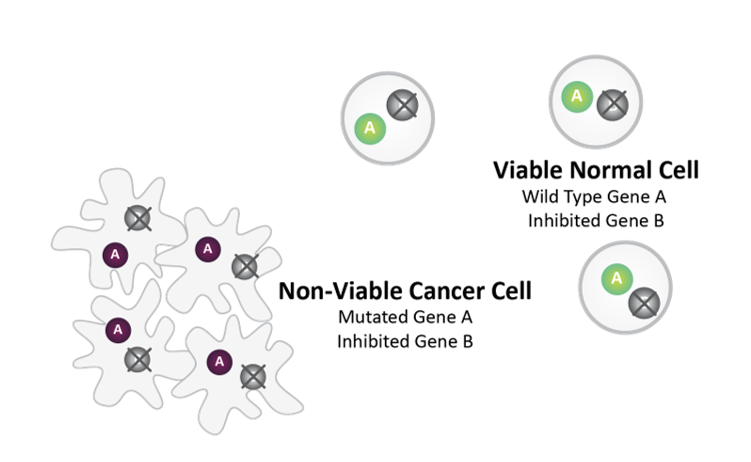
Synthetic lethality between mutated Gene A and pharmacologically inhibited Gene B causes cancer cell death.
Normal Cell survives as it does not harbor Gene A mutation.
Synthetic Lethality Target Discovery
Synthetic lethality is a core research focus for IDEAYA. We have capabilities for identification and validation of new synthetic lethality targets. For targets of interest, we advance our research to discover therapeutic drugs and relevant biomarkers.
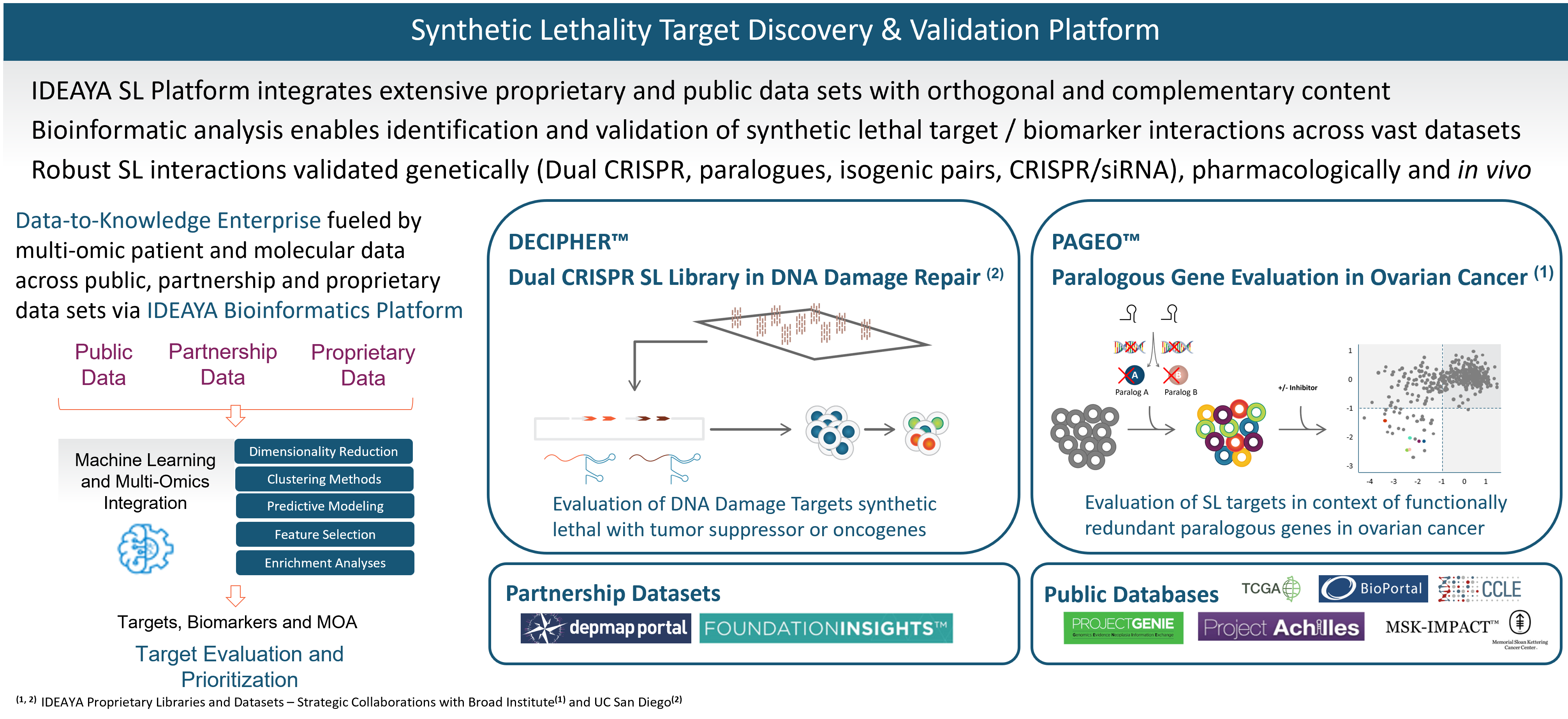
Our synthetic lethality target and biomarker discovery platform integrates a broad set of computational and functional capabilities, including synthetic lethality screening libraries, bioinformatics, and experimental validation.
Our target discovery platform includes a proprietary library and data set resulting from our DECIPHER™ Dual CRISPR Synthetic Lethality library constructed in collaboration with Dr. Trey Ideker, Ph.D., at University of California, San Diego. Synthetic lethality targets are identified through simultaneous knockout of two genes in a library of curated human cell lines. In an initial screen, one of the genes being perturbed is a known tumor suppressor gene and biomarker, and the other gene encodes a potential synthetic lethality target. We have been developing our synthetic lethality target discovery library in collaboration with Trey Ideker, Ph.D. at UCSD.
Our platform also includes proprietary data from our PAGEO™ (Paralogous Gene Evaluation in Ovarian cancer) library being developed in collaboration with the Broad Institute of Harvard and MIT. This collaboration will use the large-scale CRISPR paralog screening platform developed at the laboratory of William R. Sellers, M.D., Core Institute Member, Broad Institute, to evaluate functionally redundant paralogous genes across ovarian cancer subtypes and to generate novel target and biomarker hypotheses. Dr. Sellers, who also serves on our Scientific Advisory Board, is the principal investigator.
In addition to our proprietary PAGEO and DECIPHER data sets, IDEAYA has access to partnership data sets and public databases. We are members of the Broad Institute’s Cancer Dependency Map (DepMap) Consortium, enabling access to genome-wide screens of cell lines based on single CRISPR gene editing. We also evaluate Foundation Medicines’ FoundationInsights™. We access public databases such as Novartis’ Project Drive, Broad Institute’s Project Achilles, The Cancer Genome Atlas, or TCGA, Memorial Sloan Kettering Cancer Center’s IMPACT, American Association for Cancer Research’s Project GENIE, Cancer Cell Line Encyclopedia, Genomics England’s 100,000 Genomes Project, and National Institutes of Health’s Genotype-Tissue Expression Portal.
We have established bioinformatics capabilities, which we are applying to integrate across each of the orthogonal data sets in our platform, including using proprietary algorithms and unsupervised machine learning.
We have capabilities for developing in vitro and in vivo models for target and biomarker validation. Translational models are selected based on our synthetic lethality expertise and informed by computational methods enabled through bioinformatics.
Synthetic Lethality Target Discovery Approach
Translational Biology
IDEAYA’s translational biology platform is based on cutting-edge technologies for discovery, validation and clinical development of biomarkers in parallel with novel therapeutics. Biomarker-enabled translational approaches help enhance patient outcomes by identifying patients likely to respond to particular therapies and by providing clinical insights based on extent of target modulation.
A biomarker is a biological molecule found in tissue or bodily fluid that indicates a normal or abnormal process, or a condition or disease. Biomarkers can be, for example, DNA, RNA, protein, or metabolites.
Diagnostic biomarkers include predictive, prognostic and resistance biomarkers. For a particular cancer indication, or potentially pan-indication, predictive biomarkers help select for patients most likely to benefit from a drug. Prognostic biomarkers provide information about a patient’s overall outcome, independent of a particular therapy. Resistance biomarkers enable clinical decisions for potential combinations and/ or next-line therapies.
Pharmacodynamic biomarkers are used both in pre-treatment and post-treatment testing to confirm and understand the extent of target modulation. In addition to providing biological proof-of-activity, such information can inform clinical decision making – e.g., dosing amounts and/or schedules.
Companion diagnostics (CDx) are diagnostic products intended for launch with a therapeutic drug. CDx products help support clinical decision-making as a diagnostic or pharmacodynamic biomarkers.
IDEAYA’s translational platform includes in-house capabilities coupled with strategic networks and partnerships for discovery, validation and clinical development of biomarkers.
References
General Synthetic Lethality
- Topatana, W., Juengpanich, S, Li, S., et al. Advances in synthetic lethality for cancer therapy: cellular mechanism and clinical translation. J Hem Onc (2020) 13:118
- Kaelin WG. Synthetic lethality: a framework for the development of wiser cancer therapeutics. Genome Med. 2009;1(10):99.
- Hartwell LH, Szankasi P, Roberts CJ, et al. Integrating genetic approaches into the discovery of anticancer drugs. Science. 1997;278(5340):1064-8.
- Shen JP, Zhao D, Sasik R, et al. Combinatorial CRISPR-Cas9 screens for de novo mapping of genetic interactions. Nat Methods. 2017;14(6):573-576.
- Kaelin, W. The Concept of Synthetic Lethality in the Context of Anticancer Therapy. Nat Rev Cancer 5, 689–698 (2005).
- Huang, A., Garraway, L.A., Ashworth, A. et al. Synthetic lethality as an engine for cancer drug target discovery. Nat Rev Drug Discov 19, 23–38 (2020).
- O’Neil NJ, Bailey ML, Hieter P. Synthetic lethality and cancer. Nat Rev Genet. 2017;18(10):613-623.
- Tsherniak A, Vazquez F, Montgomery PG, et al. Defining a Cancer Dependency Map. Cell. 2017;170(3):564-576.e16.
- McDonald ER 3rd, de Weck A, Schlabach MR, et al. Project DRIVE: A Compendium of Cancer Dependencies and Synthetic Lethal Relationships Uncovered by Large-Scale, Deep RNAi Screening. Cell. 2017;170(3):577-592.e10.
- Yu C, Mannan AM, Yvone GM, et al. High-throughput identification of genotype-specific cancer vulnerabilities in mixtures of barcoded tumor cell lines. Nat Biotechnol. 2016;34(4):419-423.
- Mullard A. What’s next for the synthetic lethality drug discovery engine?. Nat Rev Drug Discov. 2022;21(7):477-479.
MAT2A
- Guo J., Yang Y., Buettner R., Rosen S. Targeting the methionine – methionine adenosyl transferase 2A – S-adenosyl methionine axis for cancer therapy. Curr Opin Oncol.2022; 34(5):546-551
- Marjon K., Kalev P., Marks K., Cancer Dependencies PRMT5 and MAT2A in MTAPp16-Deleted Cancers. Annu. Rev. Cancer Biol. 2021. 5:371-90
- Kalev, P., Hyer, Marc L., Gross, Stefan et al. MAT2A Inhibition Blocks the Growth of MTAP-Deleted Cancer Cells by Reducing PRMT5-Dependent mRNA Splicing and Inducing DNA Damage. Cancer Cell. 2020; 39,209–224
- Marjon K, Cameron MJ, Quang P, et al. MTAP Deletions in Cancer Create Vulnerability to Targeting of the MAT2A/PRMT5/RIOK1 Axis. Cell Rep. 2016;15(3):574-587.
- Mavrakis KJ, Mcdonald ER, Schlabach MR, et al. Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5. Science. 2016;351(6278):1208-13.
- Kryukov GV, Wilson FH, Ruth JR, et al. MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science. 2016;351(6278):1214-8.
DLL3
PARG
- Harrision D, Gravells P, Thompson R, et al. Poly(ADP-Ribose) Glycohydrolase (PARG) vs. Poly(ADP-Ribose) Polymerase (PARP) – Function in Genome Maintenance and Relevance of Inhibitors for Anti-cancer Therapy. Front Mol Biosci. 2020; 7:191
- Zhang M, Lai Y, Vasquez JL, et al. Androgen Receptor and Poly(ADP-ribose) Glycohydrolase Inhibition Increases Efficiency of Androgen Ablation in Prostate Cancer Cells. Sci Rep. 2020;10(1):3836.
- Pillay N, Tighe A, Nelson L, et al. DNA Replication Vulnerabilities Render Ovarian Cancer Cells Sensitive to Poly(ADP-Ribose) Glycohydrolase Inhibitors. Cancer Cell. 2019;35(3):519-533.e8.
- McDermott N, Buechelmaier E, Powell S. Capitalizing on Cancer Replication Stress by Preventing PAR Chain Turnover: A new Type of Synthetic Lethality. Cancer Cell. 2019;35:344-346.
- Pillay N, Brady R, Dey M, et al. DNA replication stress and emerging prospects for PARG inhibitors in ovarian cancer therapy. 2021;163:160-170.
- Moore G, Powell S, Higginson D, et al. Examining the prevalence of homologous recombination repair defects in ER+ breast cancers. Breast Cancer Research and Treat. 2022;192:649-653.
- Sun Y, Chen J, Huang S-Y, et al. PARylation prevents the proteasomal degradation of topoisomerase I DNA-protein crosslinks and induces their deubiquitylation. Nat Commun 2021;12(1):5010.
- Townsend Stork C, Bocek M, Crossley MP, et al. Co-transcriptional R-loops are the main cause of estrogen-induced DNA damage. eLife. 2016;5:e17548.
- Wright RH, Lioutas A, Le Dily F, et al. ADP-ribose-derived nuclear ATP synthesis by NUDIX5 is required for chromatin remodeling. Science. 2016 Jun 3;352(6290):1221-5.
POLQ
- Feng W, Simpson DA, Carvajal-Garcia J, et al. Genetic determinants of cellular addiction to DNA polymerase theta. Nat Commun. 2019 Sep 19;10(1):4286
- Mengwasser KE, Adeyemi RO, Leng Y, et al. Genetic Screens Reveal FEN1 and APEX2 as BRCA2 Synthetic Lethal Targets. Mol Cell. 2019;73(5):885-899.e6.
- Ceccaldi R, Liu JC, Amunugama R, et al. Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nature. 2015;518(7538):258-62.
- Mateos-gomez PA, Gong F, Nair N, et al. Mammalian polymerase θ promotes alternative NHEJ and suppresses recombination. Nature. 2015;518(7538):254-7.
- Belan O, Sebald M, Adamowicz M, et all. POLQ seals post-replicative ssDNA gaps to maintain genome stability in BRCA-deficient cancer cells. Molecular Cell. 2022;82(24):4664-4680.e9
- Schrempf A, Bernardo S, Verge E, et al. POLθ processes ssDNA gaps and promotes replication fork progression in BRCA1-deficient cells. Cell Rep. 2022;41(9):111716.
- Mann A, Ramirez-Otero M, De Antoni A. POLθ prevents MRE11-NBS1-CtIP-dependent fork breakage in the absence of BRCA2/RAD51 by filling lagging-strand gaps. Mol Cell. 2022;82(22):4218-4231.e8.
- Schrempf A, Slyskova J, Loizou J. Targeting the DNA Repair Enzyme Polymerase θ in Cancer Therapy. Trends Cancer. 2021;7(2):98-111.
WRN
- van Wietmarschen N, Sridharan S, Nathan WJ, et al. Repeat expansions confer WRN dependence in microsatellite-unstable cancers. Nature 2020;586(7828):292-298
- Behan FM, Iorio F, Picco G, et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature. 2019;568(7753):511-516.
- Chan EM, Shibue T, Mcfarland JM, et al. WRN helicase is a synthetic lethal target in microsatellite unstable cancers. Nature. 2019;568(7753):551-556.
- Kategaya L, Perumal SK, Hager JH, et al. Werner Syndrome Helicase Is Required for the Survival of Cancer Cells with Microsatellite Instability. iScience. 2019;13:488-497.
- Lieb S, Blaha-ostermann S, Kamper E, et al. Werner syndrome helicase is a selective vulnerability of microsatellite instability-high tumor cells. Elife. 2019;8.
Darovasertib (IDE196)
- Piperno-Neumann, S. et al A phase I trial of LXS196, a protein kinase C (PKC) inhibitor, for metastatic uveal melanoma British Journal of Cancer 2023;
- Parker, P. et al Equivocal, Explicit and Emergent Actions of PKC isoforms in Cancer Nat Rev Cancer 2021; 21(1): 51-63
- Ito, Takahiro, Young Michael J., et all. Paralog knockout profiling identifies DUSP4 and DUSP6 as a digenic dependence in MAPK pathway-driven cancers Nature Genetics 2021; 53: 1664-1672
- Watson, L. et al Co-Ordinated Control of the Aurora B Abscission Checkpoint by PKCε Complex Assembly, Midbody Recruitment and Retention Biochemical Journal 2021; 478: 2247-2263
- Garg, R. et al PKC Epsilon is Required for KRAS-driven Lung Tumorigenesis Cancer Res. 2020; 80(23): 5166-5173
- Parker, P. et al A Cancer-associated, Genome protective Programme Engaging PKCε Advances in Biological Regulation 2020; 78: 2212-4926
- Brownlow, N. et al Mitotic Catenation is Monitored and Resolved by a PKCε-regulated Pathway Nature Communications 2014; 5: 5685
- Symonds, J. et al Protein Kinase Cdelta is a downstream effector of oncogenic KRAS in lung tumors Cancer Res. 2011; 71(6): 2087-2097
Uveal Melanoma
- Shields CL, Furuta M, Thangappan A, et al. Metastasis of Uveal Melanoma Millimeter-by-Millimeter in 8033 Consecutive Eyes. Arch Ophthalmol. 2009;127(8):989–
998. - Van Raamsdonk CD, Bezrookove V, Green G, et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 2009; 457(7229):599-602
- Croce M, Ferrini S, Pfeffer U, et al. Targeted Therapy of Uveal Melanoma: Recent Failures and New Perspectives. Cancers (Basel) 2019 18;11(6):846
- Lane AM, Kim IK, and Gragoudas ES. Survival rates in patients after treatment for metastasis from uveal melanoma. JAMA Ophthal. 2018; 136(9):981-986.
- Surriga O, Rajasekhar VK, Ambrosini G, et al, Crizotinib, a c-Met inhibitor, prevents metastasis in a metastatic uveal melanoma model. Mol Cancer Ther. 2013;12(12):2817-26
- Cheng H, Chua V, Liao C, et al, Co-targeting HGF/cMET Signaling with MEK Inhibitors in Metastatic Uveal Melanoma. Mol Cancer Ther. 2017; 16(3):516-528.
- Van raamsdonk CD, Griewank KG, Crosby MB, et al. Mutations in GNA11 in uveal melanoma. N Engl J Med. 2010;363(23):2191-9.
- Beasley A, Isaacs T, Khattak MA, et al. Clinical Application of Circulating Tumor Cells and Circulating Tumor DNA in Uveal Melanoma. JCO Precision Onc 2018;(2):1-12.
- Rantala ES, Hernberg MM, Piperno-Neumann S, et al. Metastatic uveal melanoma: The final frontier. Progress in Retinal and Eye Research. 2022;(90) 101041.
- Rantala ES, Hernberg M, Kivelä TT. Overall survival after treatment for metastatic uveal melanoma: a systematic review and meta-analysis. Melanoma Res. 2019;29(6):561-568.
- Khoja L, Atenafu EG, Suciu S, Leyvraz S, et al. Meta-analysis in metastatic uveal melanoma to determine progression free and overall survival benchmarks: an international rare cancers initiative (IRCI) ocular melanoma study. Ann Oncol. 2019;30(8):1370-1380.
- Piulats JM, Espinosa E, de la Cruz Merino L, et al. Nivolumab Plus Ipilimumab for Treatment-Naïve Metastatic Uveal Melanoma: An Open-Label, Multicenter, Phase II Trial by the Spanish Multidisciplinary Melanoma Group. J Clin Oncol. 2021; 39(6):586-598.
PKC + cMET Combo
- Kermogant K, Zicha D, Parker PJ, PKC controls HGF-dependent c-Met traffic, signalling and cell migration. EMBO 2004, 23, 3721–3734
- Rosse C, Linch M, Kermorgant S, et al. PKC and the control of localized signal dynamics. Nat Rev Mol Cell Biol. 2010;11(2):103-12
- Leyvraz S, Konietschke F, Peuker C, Biomarker-driven theraphis for metastatic uveal melanoma: A prospective precision oncology feasibility study. European Journal of Cancer. 2022;169:146-155.
